Priscilla Nibi: Experience in the assessment of companion diagnostics by the EMA
A systematic review of the assessments of companion diagnostics by the EMA, in 2017-2019. Project of Priscilla Nibi, student of Utrecht University. She performed her research internship at the Medicines Evaluation Board (CBG-MEB) under supervision of Marc Maliepaard, Marjon Pasmooij and Helga Gardarsdottir.
Background
Our healthcare system is transitioning from a “one-size-fits-all” to a personalised medicine approach. In vitro diagnostics (IVDs) like companion diagnostic (CDx) have are already become indispensable in the personalised medicine approach. A CDx is a (validated) test for a predictive biomarker (PBM). Testing with a certain CDx is prerequisite to the correct and safe use of the corresponding medicine and essential before the initiation of treatment.
Up until now CDx have not been defined in European legislation and because of that these diagnostics can be certified based on their manufacturers' information only. This is risky because no proof of CDx performance is necessary for this. This will change with the implementation of two new regulations on medical devices (EU 2017/745) and IVD (EU 2017/746) on 26 May 2021 and 26 May 2022, respectively. The IVD regulation will for the first time in EU legislation specifically address CDxs.
The most important changes will be that CDxs will be classified as having high individual risk or moderate public risk (category C) and that Notified Bodies (NBs) will have to assess CDx performance. With the changing role of NBs in the assessment of CDx, the role of the European Medicines Agency (EMA) changes as well. The EMA will play the role of advisor for the clinical assessment of CDx and the EMA will have to prepare for this new role.
Main research question
How has the EMA assessed CDx in the past (2017 - 2019) and what can be learnt from this regarding the new regulation for IVDs that will be implemented in 2022?
Methods
A systematic review of previous assessments provided by the EMA on medicinal products involving CDx was carried out. An overview of all the medicines that were applied for marketing authorisation through the centralized authorisation procedure between 1995 and 2019 was downloaded from the EMA website. The investigation was restricted to medicines approved between 2017 and 2019.
The Summary of product characteristics (SmPC) and European Public Assessment Reports (EPARs) of these products were screened to select the products with a CDx. After this both the EPARs and the internal assessment reports (day 80 and day 120 ARs) of the centralised procedures were analysed to see if and how CDx were assessed. Special attention was paid to the four categories from the EMA concept paper on predictive biomarker-based assay development in the context of drug development and lifecycle: 1) analytical performance of the CDx, 2) clinical performance of the CDx, 3) interchangeability of assays, including concordance testing, bridging studies and tests for the same PBM, and 4) testing of stored or fresh patients biomarker samples.
The results for the four different topics were then compared to identify if any topic was discussed more often. Finally, the results from the different sources (EPARs and ARs) were also compared, to identify if there were any differences noticeable between the statements on PBMs and CDx.
Results
We identified 167 medicines authorised in 2017 - 2019. Initial screening indicated that a CDx was present in the EPAR for 20 of these 167 medicines (12%). More elaborate screening of the EPARs learned that of these 20 medicines, discussion of at least one of the four topics of the concept paper was presented or referred to 27 times in total. These were divided over 12 of the 20 different medicines (60%). Following the same screening approach, in the day 80 and day 120 ARs, a larger number of discussions on CDx topics (i.e., 110) were found, divided over 18 of the 20 medicines (90%). For both the EPARs and the ARs clinical performance of the CDx was discussed most often. It was mentioned 11 times in the EPARs and 59 times in the ARs.
Conclusions
To conclude, a lot of discussion about the four topics from the EMA concept paper was found in the EPARS and ARs. There was a noteworthy difference between the quantity of remarks found in the EPAR vs in the ARs, with more information on CDx being found in the ARs.
The results of this study reveal that the EMA has been assessing CDx in the past three years (2017-2019), despite that there was no regulation that required this from the EMA. The results from this study confirm the importance of the four topics from the concept paper when it comes to the clinical assessment of CDx. Especially the clinical and analytical performance of CDx were discussed thoroughly.

Fatima Tarrahi: Safety-related changes to the drug label
Project of Fatima Tarrahi, student of Utrecht University. She performed her research internship at the Medicines Evaluation Board (CBG-MEB) under supervision of Peter Mol, Viktoriia Starokozhko and Ton de Boer (Utrecht University).
For the past six months I had the opportunity to perform my research internship at the MEB, as part of the Master's pharmacy program at the Utrecht University. During this time I assessed the importance of safety data that originated from post-marketing efficacy trials in keeping the safety section of the drug label up to date. We performed this study as the International Council of Harmonization (ICH) is writing a new guideline on selective safety data collection in late stage pre-approval and post-approval efficacy trials, the ICH E19 guideline. This guideline describes that non-serious adverse events (NSAEs), and clinical lab markers that occur frequently and have been identified with sufficient precision in earlier studies, do not need to be collected in late stage clinical trials when the safety profile of the drug is reasonably well characterized. However, this also raises concern about the potential loss of important non-serious safety information from these trials. As of yet, there is no evidence that substantial safety information would be lost if the E19 concept is applied. Therefore, this study aimed to address this lack of evidence by looking at the changes in the safety section of various drug labels following late stage clinical trials.
The study population consisted of anticoagulant, antidiabetic, and lipid-lowering drugs centrally registered at the European Medicines Agency (EMA) for which a post-marketing efficacy trial was performed. The assessment histories of the European Public Assessment Reports (EPARs) were consulted, alongside previous versions of the drug labels to extract the exact changes made to the safety sections following these trials. The primary outcome described the number of changes in NSAE information. The secondary outcome characterized all the changes made to the safety sections of the included drug labels.
At the start of this study, 1,667 drugs were centrally registered at the EMA, as shown in figure 1. In total, 24 drugs met the inclusion criteria, with one or more post-approval trials supporting the variation procedure to change the drug label. Altogether, 44 trials were mentioned in these variation procedures, including in total 343,422 patients. About half (n=24) of the trials led to at least one change in the safety section, with a total number of 47 changes. Specifically, only five of the included trials led to a total of 16 (34%) changes in the NSAE information, of which six (12.8%) concerned a change of frequency, and 10 (21.2%) new NSAEs were added. More than half (n=24, 51.1%) of the changes in the safety section were categorized as other changes. These were updates of the text describing the bigger overall safety population pool. Furthermore, three (6.4%) new serious adverse events were found, and four (8.5%) frequency changes of known serious adverse events were observed.
To highlight the primary outcome, only five of the 44 included trials led to a change in NSAE information. In the selected population of anticoagulant, antidiabetic, and lipid lowering drugs, a total of 343,422 patients were needed to find sixteen changes in NSAE information, Thus, the process of comprehensive safety data collection in the selected population did not yield substantial amounts of new non-serious safety information. Based on this finding, it may be concluded that applying selective safety data collection would not lead to a substantial loss of non-serious safety information. However, it also needs mentioning that more research in this field is necessary to provide robust evidence that could support the ICH E19 draft. Implementation of selective safety data collection following the ICH E19 concept could ideally lead to an increased efficiency of performing post-marketing efficacy trials, whereas safety information in the drug label would still be reliable with most of this information originating from other sources (safety trials, periodic safety assessments) of safety documentation already in use.
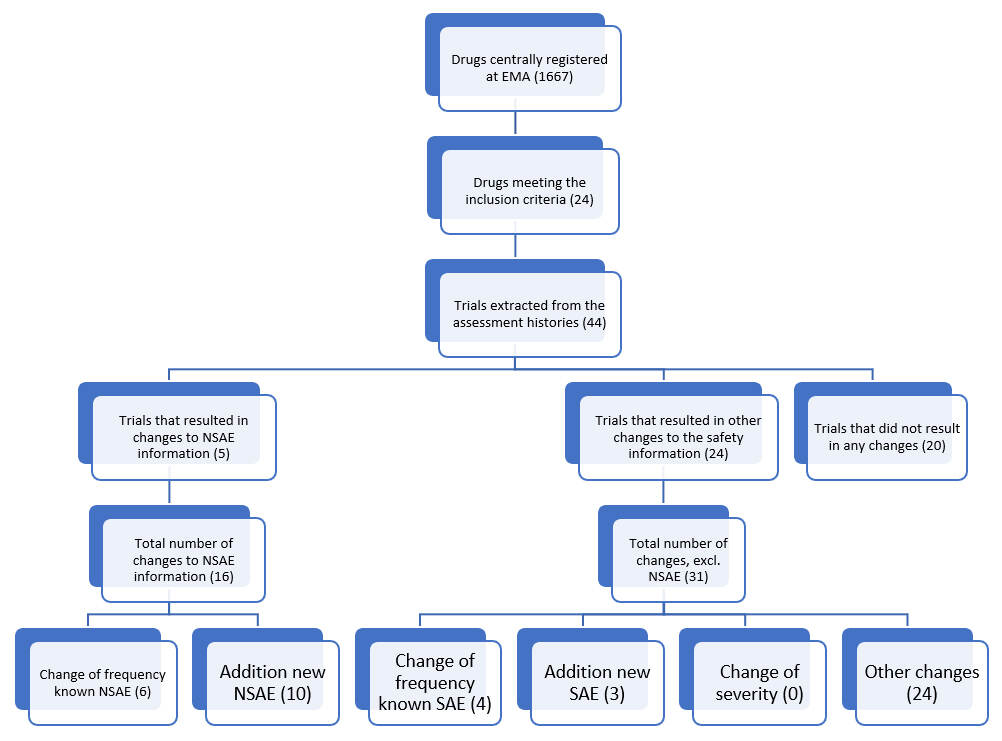
Drug centrally registered at EMA (1667)
Drugs meeting the inclusion criteria (24)
Trials extracted from the assesment histories (44)
> Trials that resulted in changes to NSAE information (5)
- Total number of changes to NSAE information (16)
-- Change of frequency known NSAE (6)
-- Addition new NSAE (10)
> Trials that resulted in other changes to the safety information (24)
- Total number of changes, excl. NSAE (31)
-- Change of frequency known SAE (4)
-- Addition new NSAE (3)
-- Change of severity (0)
-- Other changes (24)
> Trials that did not result in any changes (20)