Nienke Sikkes: Advanced melanoma patients with brain metastasis in clinical trials vs the real world
In September I started with my research project at the Medicines Evaluation Board (MEB). During the master phase of the study pharmacy we get the opportunity to do a research internship for 5 months. I chose to do this internship at the MEB because of an interesting research project in collaboration with the Dutch Institute for Clinical Auditing (DICA). Further, it provided me the opportunity to obtain a better overview of the regulatory side of medicine.
My research project focusses on the comparability of the efficacy of immuno- and targeted therapies in advanced (stage IIIc and IV) melanoma patients with brain metastasis participating in clinical trials compared to patients treated with these agents in the real world.
Patients with brain metastasis are often excluded from clinical trials because of their poor prognoses. However, melanoma is one of the most common cancers to metastasize to the brain. According to autopsy data up to 70 to 80% of metastatic melanoma patients have brain involvement. In clinical practice this results in advanced melanoma patients with brain metastasis who are treated with novel immuno- and targeted therapies by medical oncologists, despite limited available data to support their clinical decision making. Advanced melanoma patients with active brain metastases or a poor performance score status (2 or more) account for 74% of the patients treated in the real world, whereas they are non-eligible for immuno- and targeted clinical trials. This creates an unmet medical need for more data to treat these patients effectively.
Recently some post-approval studies have focussed on this specific group of patients, providing clinical practitioners data to support their clinical decision making. However, we do not know if these strict clinical studies reflect patients treated in the real world. By investigating the comparability of the efficacy outcomes of the post-approval studies with efficacy outcomes of the Dutch Melanoma Treatment Registry (DMTR), we hope to provide a better insight in differences or similarities between patients and clinical outcomes in clinical trials and the real world. The main goal will be to provide the MEB with feedback on how clinical trials reflect the real world patients. This research project was set up in line with the PhD project of Rawa Ismail.
More information on Rawa’s PhD project can be found in the previous issue of Regulatory Science Magazine.

Rosalinde Bot: Development of clinical value over the life cycle of oncology medicines in Europe: a comparison of conditionally and standard approved medicines
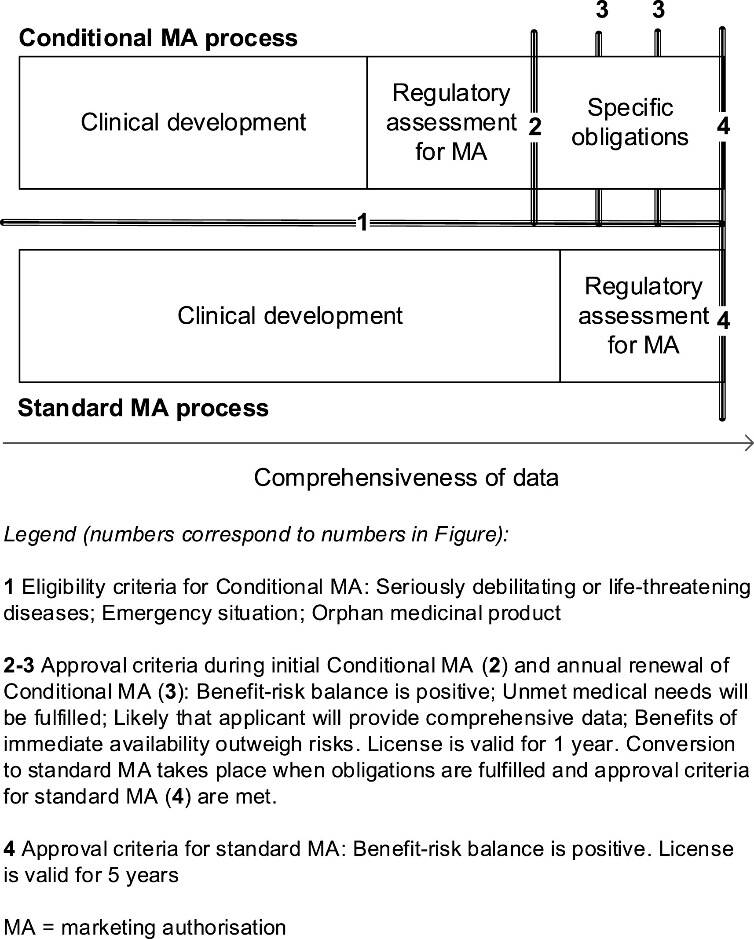
Conditional marketing authorisation (CMA) is a pathway for innovative medicines to enter the market in Europe earlier than through the ‘standard’ authorisation pathway. These medicines are approved based on less comprehensive data but need to fulfil (additional) post-approval studies, named specific obligations. When these studies are fulfilled, the conditional authorisation will be converted to a standard authorisation, provided that uncertainties are sufficiently resolved and the benefit-risk is still positive. The pathway of CMA, compared to the standard pathway, is shown in figure 1.
The clinical value of medicines is perceived in clinical practice. Medicines with a CMA are approved with a promise of innovation, but less comprehensive data may lead to less understanding of the clinical value in the clinical practice. For this reason, we studied the impact of CMA on the clinical value of medicines. Since more than 50% of all conditional approvals are oncology medicines, this project aimed to investigate the development of the clinical value of conditionally versus standard approved oncology medicines.
For this project, all conditionally approved oncology medicines that were converted to a standard marketing authorisation before or on the 28th of February 2019 were identified (n=14). To complement them, standard approved oncology medicines that were similar with regard to pharmacotherapeutic group and indication were identified (n=22).
The European Society for Medical Oncology Magnitude of Clinical Benefit Scale (ESMO-MCBS) is a validated scale to give a score to a clinical study. These scores represent the clinical value of the medicine. Studies with endpoints overall survival, progression-free survival, time to progression and objective response rate are eligible for this scale. In this research, clinical value was determined based on pivotal studies underlying 1) initial approval, 2) modifications of the initial indication, and 3) new indications. For conditionally approved medicines, clinical value was also determined at conversion to standard authorisation, based on specific obligation studies.
Two methods were used to compare the clinical value of the conditionally and standard approved medicines during the drug life cycle. First of all, the ESMO-MCBS score of the medicines was determined (score 0-5) and secondly, the amount of medicines with substantial clinical value (score 4-5 versus 0-3). The clinical value of conditional and standard approvals was compared at four moments in the drug life cycle: 1) at conversion for conditionally approved versus at initial approval for standard approved medicines, 2) at modifications of the initial indication, 3) at new indications, and 4) overall, combining all of the above. For all these moments, the highest score for each medicine, available at that moment, was selected for the comparison. In figure 2 and 3, comparison number 1 is shown for both methods.
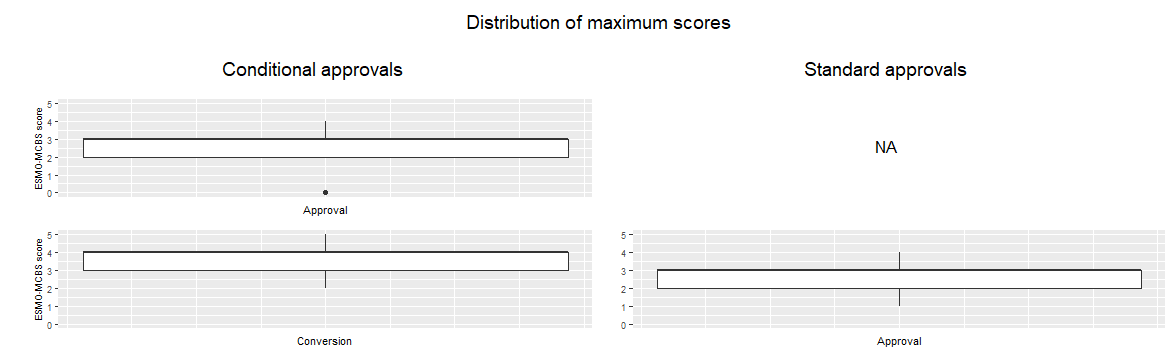
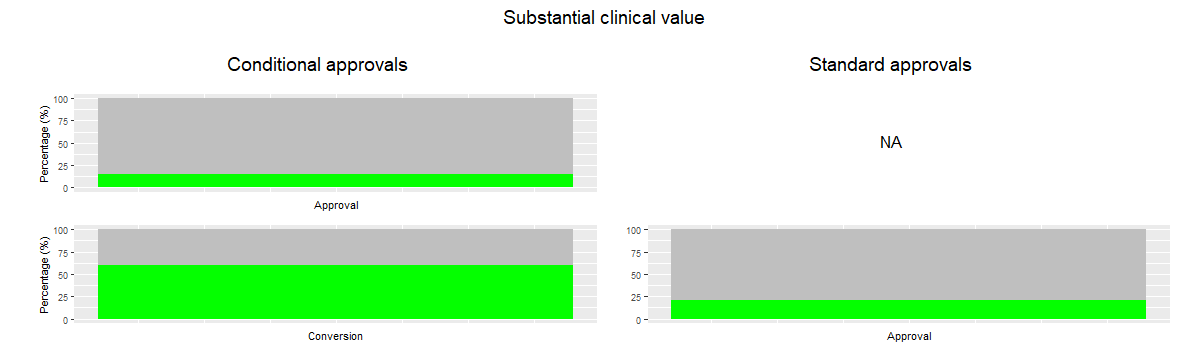
These two methods showed similar results. At approval, the conditional approvals have the same clinical value as the standard approvals, but when the specific obligations are submitted and fulfilled, the clinical value is higher for the conditional approvals than for the standard approvals. The amount of medicines with substantial clinical value is higher for the conditional approvals as well. When assessing the modifications of the initial indication, the conditional approvals score again higher than the standard approvals.
Standard approvals had more extensions of indication than the conditionals approvals. Extensions of indications of conditional approvals had a lower clinical value than the standard approvals. When including all indications in the analysis, conditionally approved and standard approved medicines had a similar overall clinical value.
In conclusion, this study showed that, when assessing the initial indication, the conditional approvals have a higher clinical value than the standard approvals, but when all indications are included, the clinical value is similar.
This study provides new information on the functioning of the conditional marketing authorisation pathway. It shows that specific obligations give the needed comprehensive data to increase the knowledge of the clinical value of medicines over time. As a follow-up, analyses of specific cases could inform the regulatory system on best and lesser practices to achieve knowledge on clinical value. This may help to optimise the use of CMA (for oncology medicines) to provide early access to highly innovative medicines. In the end, this will lead to a better treatment for the patients, since these can get new innovative medicines with a high potential earlier.
Source
1. Hoekman, J. et al. Use of the conditional marketing authorization pathway for oncology medicines in Europe. Clin. Pharmacol. Ther. 98, 534–541 (2015).