A variety of PhD projects are ongoing with involvement of the MEB in close collaboration with academic partners and institutes. Below an overview is presented of all these PhD projects. Involvement varies from PhD projects that are funded by the MEB in which PhD students work about 50% of the time as assessor or regulatory project leader at the MEB and 50% of their time on their PhD project at the University, to PhD projects where MEB involvement is limited to collaboration of MEB employees on specific chapters of the PhD thesis.
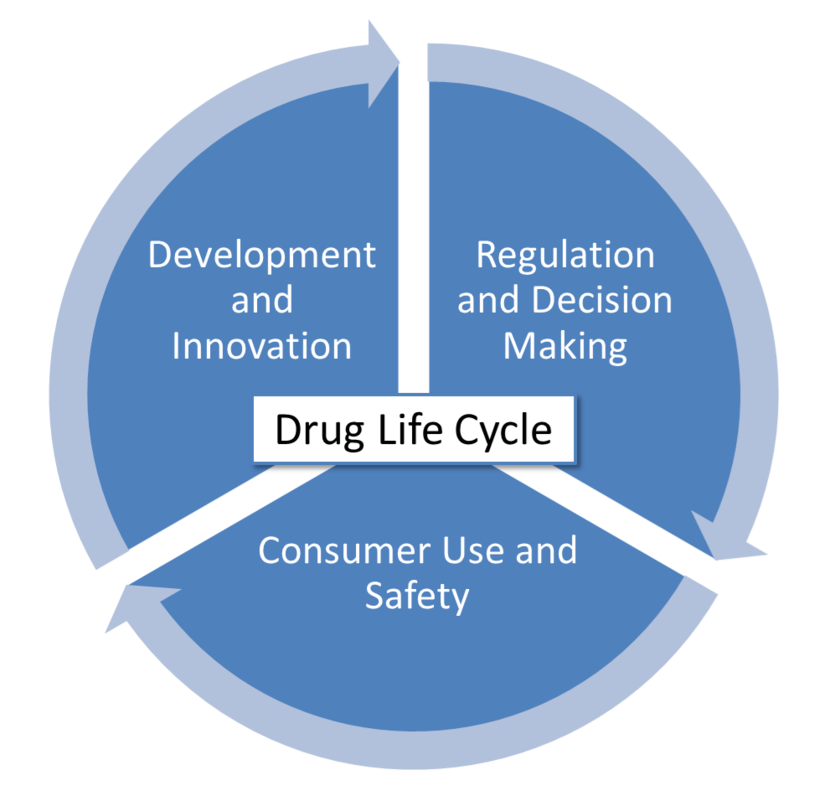
The projects are divided in three domains related to the drug life cycle:
- Development and Innovation
- Regulation and Decision Making
- Consumer Use and Safety
Overview of PhD projects
Domain 1: Development and Innovation
PhD-student: Coen Stalpers (MEB / Dutch National Institute for Public Health and Environment (RIVM))
Promotors: Prof. Willem van Eden (Utrecht University) and Prof. Femke Broere (Utrecht University)
Co-promotors: Dr. Rob Vandebriel (RIVM) and Dr. Marcel Hoefnagel (MEB)
The research is part of the IMI project VAC2VAC which aims to develop, optimise and evaluate physico-chemical and immunochemical methods, cell-based and other assays for routine batch quality, safety and efficacy testing of vaccines (http://www.vac2vac.eu/). This PhD project is focussed on the development of in vitro alternatives for the use of test animals in lot release testing of the Diphtheria, Tetanus and acellular Pertussis combination vaccines (DTaP). To ensure product quality, each produced DTaP lot is tested by the manufacturer and the OMCL (Official Medicine Control Laboratory) for release to the EU market. Safety and efficacy testing involves the use of many mice and Guinea pigs. Drawbacks of animal tests are that results are highly variable, furthermore, animal tests are time consuming, expensive and raise moral concern. In the lab we spend time performing experiments with cultured immune cells in an attempt to develop an in vitro assay that may play a part in an animal free potency testing strategy for DTaP. The aim of this research is to contribute to development of innovative testing methods that will replace animal tests, and will also help to formulate scientific arguments to guide regulatory decision-making concerning the use of animal free potency testing methods.
PhD-student: Britt Duijndam (MEB / Leiden Academic Centre for Drug Research, Leiden University)
Promotor: Prof. Bob van de Water (Leiden University)
Co-promotor: Dr. Jan Willem van der Laan (MEB)
Nuclear hormone receptors (NHRs) regulate gene expression networks involved in various biological processes, such as cell growth, proliferation and homeostasis. Disruption of these networks, for instance with non-genotoxic carcinogens, can result in adverse outcomes such as unanticipated cell proliferation ultimately culminating in tumor formation. Since NHR signaling is also involved in normal physiological responses, and not solely activated in adverse outcomes, safety assessment in the context of NHR activation remains difficult. For this reason, it is essential to define quantitative relationships between different key events leading to a particular adverse outcome induced by non-physiological NHR activation. We established fluorescent protein reporter cell lines of important players in the NHR signaling pathway in human relevant cell lines. These in vitro reporters enable to monitor the following key events: receptor translocation/activation, downstream transcriptional activation and cell cycle progression, leading up to proliferation. In combination with advanced live cell imaging, these reporters can monitor the spatial and temporal dynamics of these key events at a single cell level. This adverse outcome pathway (AOP)-driven reporter platform will allow us to determine the quantitative relationships between the various different key events and the ultimate cellular adverse outcome. Eventually, these reporters can be used to screen e.g. drug candidates or other chemicals of concern for the potential of modulating NHR activity and likely non-genotoxic mode of action. In this way, this study makes a first step towards an improved strategy to predict carcinogenic potential of drugs or other chemicals in humans.
PhD-student: Guilherme Ferreira (Utrecht Institute for Pharmaceutical Sciences (UIPS), Utrecht University)
Promotors: Prof. Huub Schellekens (Utrecht University) and Prof. Ellen Moors (Utrecht University)
Co-promotor: Dr Peter van Meer (MEB / Utrecht University)
The high attrition rate in drug development has been partially attributed to the poor translation of animal data to the clinic. While standards for safety evaluation are clearly defined, the assessment of efficacy is not standardised, which can arguably lead to the use of suboptimal animal models. The first project of this PhD project aimed at developing a framework to assess, validate, and compare animal models used in the preliminary assessment of efficacy dubbed FIMD (Framework to Identify Models of Disease). This project will be followed by a systematic review and meta-analysis of animal studies which will include a comparison of effect sizes of drugs in animals and humans, and the creation of a translational table for a more objective comparison of animal models within the same indication will complement FIMD. Finally, we will analyse marketing authorisation applications and submissions to the Central Committee on Research Involving Human Subjects (CCMO) to identify the justification of choice for efficacy models, and assess the robustness of the studies used to base the ‘go decision’ to clinical trials, exploring the interaction between the two institutions, and opportunities for improvement. Ultimately, the tools developed in these projects will lead to a more evidence-based selection of efficacy models, which can then allow the choice of more relevant models and therefore, improve the translation of animal to clinical data.
PhD-student: Désirée Veening-Griffioen (Utrecht Institute for Pharmaceutical Sciences (UIPS), Utrecht University)
Promotors: Prof. Huub Schellekens (Utrecht University) and Prof. Ellen Moors (Utrecht University)
Co-promotor: Dr Peter van Meer (MEB / Utrecht University)
We study the value of animal models of efficacy in drug development, since clinical failure in drug development is partly attributed to the poor translation of animal data to the clinic. One reason is poor external validity, the generalization of the results obtained in the model to the human disease.
The first project is aimed at animal models for Alzheimer’s disease, where the clinical attrition rate is more than 99%, the biology of the disease is insufficiently understood, and the currently registered drugs are not able to cure the disease. Can we identify animal models that are able to predict correctly for clinical outcome?
The next project focuses on two animal models for Cow’s Milk Allergy. For claims related to the reduction of risk to allergy to milk proteins, the formulae administered orally should not induce sensitization in animals, to the intact proteins from which the formulae are derived (Directive 96/4/EC). Can we differentiate between these models? Finally, Dutch Animal Experimentation Project licenses will be evaluated on the applicants justification for animal model choice, as well as the review by stakeholders in the application process. The data obtained in this project will facilitate the choice and justification of better translational animal models.
PhD-student: Renske ten Ham (Utrecht Institute for Pharmaceutical Sciences (UIPS), Utrecht University)
Promotors: Prof. Olaf Klungel (Utrecht University) and Prof. Bert Leufkens (Utrecht University)
Co-promotors: Dr. Anke Hövels (Utrecht University) and Dr. Jarno Hoekman (Utrecht University)
Collaborators: Dr. Marcel Hoefnagel (MEB) and Dr Hans Ovelgönne (MEB)
Cell therapy Medicinal Products (CTs) hold great promise in treatment of rare and high burden diseases. Currently, CTs on the market are priced up to €1.000.000. These prices raise concerns for healthcare access and potentially have large impact on healthcare budgets. In literature few CT cost-effectiveness analyses have been published comparing treatment benefits and costs. Yet, no transparent cost calculations or methods are available for the products themselves. Identifying the cost drivers and effect of manufacturing choices, will help value these products from developer-, investor- and payer-perspective. Costing methodologies are available for non-CTs. However, these are unsuitable for CTs due to novel manufacturing technologies and an unconventional patient-product supply chain.
Therefore, the primary aim of this research is to develop a costing framework and corresponding methodology specifically for small scale CT manufacturers. This framework may serve as a tool for manufacturing cost estimations to guide strategic decision making. To do so a multi-center costing study will be performed, in which nine different CTs will act as case-studies.
Three cell facilities across Europe will share their manufacturing data. A costing study will be performed based on Activity-Based-Costing methodology, which will be modified to fit the CT manufacturing process. The starting point is routine manufacturing and endpoint release of end-product. Unit of measure is cost/treatment (2018 EUR) using a hospital perspective. Costs will be divided in personnel, facility, equipment and materials.
Sensitivity analyses and scenarios will test cost calculations and framework for robustness. Product costs will be reported in aggregated form. We aim to make recommendations around cost drivers and possible savings for future and current CT manufacturers, regulators, payers and investors.
Medicinal product development consists of more than manufacturing. This study is a first step towards transparency in CT manufacturing cost and cost drivers. Cost of R&D and set-up of routine manufacturing is out of scope for this study but are of interest for future research, especially for pricing purposes.
Domain 2: Regulation and Decision Making
Carla Jonker (MEB / Julius Center at the Utrecht Academic Medical Centre, Utrecht University)
Promotor: Prof. Arno Hoes (Utrecht University)
Co-promotors: Dr. Marijke van den Berg (Utrecht University) and Dr. Peter Mol (MEB / University of Groningen)
Registries are, increasingly, requested by regulatory authorities to monitor safety and efficacy. The data that are requested vary with disease and product. This variability in data collection may hamper the usefulness of registries, i.e. their contribution to benefit/risk assessment over the drug life-cycle.
In this PhD project we reviewed the European Public Assessment Reports for new medicines approved in the period 2007 up to 2010. We investigated the frequency, the type, and the reason for requiring a registry. The majority of registries required by regulators were existing disease registries. We saw that registries are an important and frequently used tool for post‐approval data collection for orphan and innovative drugs. In addition, we studied the follow-up of these registries 5 years after market access. The conclusion was that enrolment of patients into post-approval registries was poor, although the results for imposed registries (these registries were a specific obligation to address a particular concern with respect to either safety or efficacy) seemed to be better. We are now in the process: 1) to compare the data from a single-arm clinical trial and a registry, the PedNet registry, in the field of haemophilia; 2) to set up a survey.
When anticipating on the requirements for registries, it is worthwhile to start building registries in the pre-licensing period, and organize them so that they are useful for multiple purposes, clinicians, industry and regulatory authorities. The goal is that regulatory authorities will better know what to learn from registries.
PhD-student: Jorn Mulder (MEB / Utrecht University)
Promotors: Prof. Ton de Boer (MEB / Utrecht University) and Prof. Emile Voest (Netherlands Cancer Institute, Amsterdam)
Co-promotors: Dr. Violeta Stoyanova (MEB) and Dr. Marjon Pasmooij (MEB)
Personalised medicine has become a frequently discussed topic in drug development and regulatory science. An example of personalised medicine is the treatment of a subgroup of patients bearing a similar characteristic (e.g. BRAFV600 mutation), instead of the ‘‘one size fits all’’ approach. Unfortunately, identifying a biomarker that enables the detection of patients that benefit most from treatment is not always straightforward. For instance, the first tissue agnostic therapies have recently been approved by the US Food and Drug Administration. Instead of focusing on only one tumour type, these therapies are biomarker-driven and can be used for numerous tumour types that are biomarker-positive. However, data regarding the benefit of such a drug might be limited or absent in several rare tumour types, making regulatory decision-making more difficult.
The aim of this project is to investigate a selection of emerging issues in the context of personalised medicine, their implications for regulatory science in general and more specifically for drug regulation. Issues are, although not limited to, basket trials and implications of the use of biomarkers. The outcome of this research will contribute to a better understanding of personalised medicine in regulatory science and will provide conclusions that will facilitate decisions in European medicines regulation with regard to personalised medicine.
PhD-student: Lourens Bloem (MEB / WHO Collaborating Centre for Pharmaceutical Policy and Regulation, Utrecht University)
Promotors: Prof. Aukje Mantel-Teeuwisse (Utrecht University) and Prof. Olaf Klungel (Utrecht University)
Co-promotors: Dr. Menno van der Elst (MEB) and Dr. Jarno Hoekman (Utrecht University)
The aim of this PhD project is to understand regulatory handling of knowledge, uncertainties and acceptable risks at approval of medicines. Specific focus is on these levels for medicines that receive early approval by e.g. conditional marketing authorisation versus those that receive regular approval. Furthermore, the project aims to assess the added value of post-approval data requested by regulators to address uncertainties and risks, and to determine the possible consequences of accepted uncertainties and risks. A first study, for instance, longitudinally assessed whether and how post-approval data requirements changed for all conditionally approved medicines up to 2016, and whether factors could be identified that were associated with change. See also Publication Highlights.
This project will provide the MEB with insight in how optimal regulatory learning in a context of limited data and (often) unmet medical need could take place and with feedback as to whether current regulatory practices aiming to do so seem to suffice. It will do so by identifying which data requests can possibly address remaining uncertainties during the post-approval phase and which mechanisms of data collection yield useful data. As a consequence, it will also identify which type of uncertainties seem less likely to be sufficiently addressed post-approval and may therefore need to be addressed pre- or upon approval.
PhD-student: Marian Mitroiu (MEB / Julius Center for Health Sciences, Utrecht University)
Promotor: Prof. Kit Roes (Utrecht University)
Co-promotors: Dr. Steven Teerenstra (MEB) and Dr. Katrien Oude Rengerink (MEB)
The revision of ICH E9 guideline ‘Statistical Principles for Clinical Trials’ aims to deal with the concept of estimands, literally: what has to be estimated. The estimand is described by the endpoint, population, population-level summary and strategy/ies to account for intercurrent events such as treatment discontinuation, treatment switch or intake of rescue medication.
E9(R1) aims to provide a framework to agree on estimands, and ensure that this leads to consistency in primary objectives of a trial, the target population, the trial design and the summary statistics. Thus, it can provide a more precise estimate for the treatment effect in relation with the clinical question of interest. It is expected to have substantial impact on the design and conduct of clinical trials (i.e. strategy for outcome data collection, methods to integrate intercurrent events more efficiently in the analysis) as well as on scientific evaluation and regulatory approval (different estimands from the same trial likely to be of different interest to involved stakeholders).
We are conducting research on clinical trials from different therapeutic areas, to gain insights in which concrete set of estimands are relevant for the different stakeholders involved. Furthermore, we are investigating efficacy analyses methods and consistency of the different estimands by application of current estimand suggestions on real life clinical trial data.
PhD-student: Sonia Roldan Muñoz (University Medical Centre Groningen)
Promotor: Prof. Hans Hillege (MEB / University Medical Centre Groningen)
Co-promotors: Dr. Peter Mol (MEB / University Medical Centre Groningen) and Dr. Douwe Postmus (University Medical Centre Groningen)
This PhD project is part of the PROMINENT program (www.prominent.umcg.nl), a four years program composed of 16 international PhD students who will work on personalised medicine in diabetic disease management. The projects within the PROMINENT program go across all different aspects of personalised medicine from disease mechanisms and drug development to the regulation and good utilization of medicines.
The aim of this PhD project is to integrate patient preferences into regulatory decisions.
We are currently exploring the factors that influence patient choices at the time of starting or continuing a treatment. The first project is focused on pregnant women’s beliefs about medication and on how those beliefs are related to the use of medication during their pregnancy. Our second project is a study among type 2 diabetic patients that evaluates which patient- and disease- related factors have an impact on the importance that patient attach to drug characteristics. We use data collected in Dutch and Turkish patients for that.
We will use different statistical models (e.g. DCE and MCDA) to quantitatively weigh the importance that patients give to the different drug effects. Ideally, we will be able to develop drug information materials that meet patient needs and we will provide feedback to regulators which outcomes matter the most to patients.
PhD-student: Pieter Glerum (Maastricht University)
Promotors: Prof. Kees Neef (Maastricht University) and Prof. David Burger (Radboud University))
Co-promotor: Dr. Marc Maliepaard (MEB)
This PhD project focusses on the interchangeability of generic medicines. One part of the project is a non-parametric pharmacokinetic modeling and simulation approach to challenge the robustness of bioequivalence. The model building and model validation has been performed and is published, the simulation part is still to come. In the other part of the PhD project we investigate how problematic the issue of generic interchangeability really is, or is not. We quantified the actual number of patients switching between equivalent drug products in the Netherlands and are relating the number of substitution induced Adverse Drug Reactions to it. This work is performed in cooperation with the the Netherlands Pharmacovigilance Centre Lareb. The overall aim of the PhD project, and benefit to regulatory decision-making, is that we build trust in the registration and use of generics, by challenging bioequivalence, which is the cornerstone of generic registration and evaluating practical issues on generic use in practice.
PhD-student: Jeroen Koomen (MEB / University of Groningen)
Promotor: Prof. Hiddo Lambers Heerspink (University of Groningen)
Co-promotors: Dr. Jasper Stevens (University of Groningen) and Dr. Peter Mol (MEB / University of Groningen)
Dose selection for a new drug intended for the treatment of type II diabetes mellitus (T2D) is typically based on the effects of the drug on glycaemic parameters. For example, dose selection of the SGLT2 inhibitors was based on the drug effects on urinary glucose excretion and for GLP-1 receptor agonists on Hba1c. However, drugs have multiple effects that can affect renal or cardiovascular outcome as much or even more than the on-target glycaemic effects. This PhD project aims to quantify dose-exposure-response relationships between drugs intended for the treatment of T2D and renal or cardiovascular response parameters. This way, we can evaluate the current dosing regimens and personalise the treatment of patients with T2D.
In the final project of the PhD thesis, the dose justification process at initial marketing authorisation will be evaluated for previously registered oral glucose-lowering drugs. The aim is to provide recommendations and considerations that need to be taken into account when selecting a dosing regimen for a new drug intended for the treatment of T2D. This will aid the regulatory decision-making process and the assessment of a to-be registered dosing regimen.
PhD-student: Rawa Ismail (Dutch Institute for Clinical Auditing / Utrecht University)
Promotor: Prof. Ton de Boer (MEB / Utrecht University)
Co-promotors: Dr. Michel Wouters (Dutch Institute for Clinical Auditing / Netherlands Cancer Institute), Dr. Doranne Hilarius (Dutch Institute for Clinical Auditing / Rode Kruis Ziekenhuis) and Dr. Maaike van Dartel (MEB)
Within the context of this PhD research, a collaboration between the MEB and the Dutch Institute for Clinical Auditing (DICA) is realized. DICA is a not-for-profit organization that facilitates the development and maintenance of national outcomes registries around medical conditions. DICA has established over 20 registries on mainly surgical and oncological conditions, such as melanoma, lung cancer and colorectal cancer. Real world data are collected in the registries to improve quality of care, by mainly doing research with these data.
The data used in this research project are collected in the Dutch Melanoma Treatment Registry (DMTR). In the past years, multiple new immunotherapies and targeted therapies are authorised for the treatment of melanoma patients. These treatments are registered in the DMTR and show a prolonged overall-survival in melanoma care, which was also shown in marketing authorisation studies. However, patients and treatment lines used in daily practice differ from the randomized controlled trials, because of the homogenic study population and treatment.
Therefore, the focus of this research will be on the differences in effectivity and toxicity of immunotherapy and targeted therapy between marketing authorisation studies and the real-world data collected by DICA in the treatment of melanoma. For example, one of the questions will focus on the patient- and tumour characteristics of long-term survivors on BRAF- and MEK-inhibitors. In another project re-challenge with targeted therapy will be investigated. This kind of information is limited in RCTs, while the heterogenic real-world population is registered in the DMTR. This real-world evidence could be helpful in future scientific advices and in the assessments of similar medicinal products by the MEB.
Domain 3: Consumer Use and Safety
PhD-student: Esther de Vries (MEB / University Medical Centre Groningen)
Promotor: Prof. Petra Denig (University Medical Centre Groningen)
Co-promotor: Dr. Peter Mol (MEB / University Medical Centre Groningen)
Regulators communicate about drug safety issues to minimise potential risk of drug harm. Currently, regulatory communication strategies do not always result in recommended changes in clinical behaviour by health care professionals (HCPs) that may reduce this risk. Earlier work, showed more severe safety issues led to more uptake of risk communication and messages aimed at specialist prescribers to lesser uptake. This PhD-project, therefore, focusses on the uptake of risk communication messages in the hospital setting as they are also the target of most risk communication by regulatory authorities and industry. To improve these communication strategies, it is important that they are attuned to HCPs’ needs, and work practice.
We aim to identify whether and how current use of direct professional healthcare communication (DHPC) in hospital setting can be improved. We also aim to develop and apply valid effect measures to study the impact of safety information, such as DHPCs.
In addition, we will also study the coverage of safety issues communicated by the authorities by newspaper and social media. This will provide us with an understanding of the possible impact lay media have on the uptake of risk communication.
Overall, a better understanding of the opinions of HCPs in hospital setting and how drug safety information is received and handled by them, can help effective communication by regulatory authorities and industry.
PhD-student: Nafise Ghalandari (Erasmus Medical Centre Rotterdam)
Promotors: Prof. Mieke Hazes (MEB / Erasmus Medical Centre Rotterdam) and Prof. Eugene Puijenbroek (LAREB / University of Groningen)
Co-promotors: Dr. Ineke Crijns (MEB) and Dr. Radboud Dolhain (Erasmus Medical Centre Rotterdam)
Biologicals are important medicine for the treatment of chronic disorders. Various chronic conditions begin in adolescence, which implies the administration of biologicals in women of childbearing age. This PhD project focusses on the safety of biological use during pregnancy/lactation in patients with chronic autoimmune diseases. Usually data on the effects of intrauterine/lactation exposure is only available in animals. As a result, most biological SmPCs have no information about human use during pregnancy/lactation and this is reported as missing information in the RMP (Risk Management Plan). We will analyse the data after it is collected in cohorts of pregnant patients with chronic rheumatic diseases and inflammatory bowel diseases. Furthermore, retrospective research will also be conducted using the pharmacovigilance databases.
The purpose of this study is to gain knowledge about the effects on offspring after exposure during pregnancy / lactation. From regulatory point of view, this can lead to a better perspective about the benefit risk balance of treatment with biologics during pregnancy/lactation and therefore their authorisation for these group of patients.
PhD-student: Doerine Postma (Utrecht University / KNMP)
Promotors: Prof. Bert Leufkens (Utrecht University) and Prof. Peter de Smet (Radboud University Nijmegen)
Co-promotors: Prof. Aukje Mantel- Teeuwisse (Utrecht University) and Dr. Kim Notenboom (MEB)
Medicines shortages are a threat to society at times that medical demands are high. The various reasons for shortages as well as its consequences need thorough investigation to identify potential solutions within the regulatory framework. The objective of the PhD project is to get more insight in the reporting of shortages, the impact on patients and risk factors for the appearance of a shortage. This insight will be achieved through the analysis of the known shortages by authorities and pharmacy practice in The Netherlands from 2012. Based on these findings, risk factors as well as models can be extracted to predict the impact of shortages. The impact of shortages on patients needs to be mitigated to meet medical demands.
Since January 1st 2017 medicines shortages have to be reported in The Netherlands to the Medicine shortages and defects notification centre of the MEB and the Health and Youth Care Inspectorate. More insight in the reporting of shortages, the impact on patients and causes can help in supplying adequate measures to anticipate on a shortages in order to reduce the impact on patients.
PhD-student: Lotte Minnema (MEB / Utrecht University)
Promotors: Prof. Bert Leufkens (Utrecht University) and Prof. Toine Egberts (Utrecht University)
Co-promotors: Dr. Thijs Giezen (MEB / Spaarne Gasthuis) and Dr. Helga Gardarsdottir (Utrecht University)
Biopharmaceuticals are important treatment options for a variety of chronic and sometimes life‐threatening diseases. Compared with the traditional small molecule drugs, biopharmaceuticals have specific characteristics, which might also influence their safety profile. Previous research that was performed in collaboration with the Medicines Evaluation Board and Utrecht University addressed several challenges of pharmacovigilance of biopharmaceuticals. The current PhD project aims to provide further insight into these challenges by studying underlying mechanisms of biopharmaceutical related adverse drug reactions and safety related label changes. The research project aligns with current needs of the EMA/MEB on evidence generation regarding safety of biopharmaceuticals. Within this scope, we assessed neuropsychiatric side effects of monoclonal antibodies following discussions on the potential risk of suicide during the assessment procedure of brodalumab (interleukin-17-inhibitor).
PhD-student: Remy Francisca (MEB / Erasmus Medical Centre, Department of Medical Informatics)
Promotor: Prof. Miriam Sturkenboom (Utrecht Medical Centre Utrecht, Julius Centre)
Co-promotors: Dr. Sabine Straus (MEB / Erasmus Medical Centre) and Dr. Inge Zomerdijk (MEB / Erasmus Medical Centre)
Risk management planning has become an integral part of proactive, life cycle approach to pharmacovigilance. The European Union Risk Management Plan (EU-RMP) is a mandatory part of the authorisation dossier for medicinal products licensed in the European Union. The EU-RMP consists of the safety specification, which describes important risks related to use of the medicinal product as well areas of missing information that might impact the benefit-risk balance; the pharmacovigilance plan, which describes the activities intended to further characterise the safety profile; and the risk minimisation plan, which describes the measures to minimise the risks associated with use of the product. Routine risk minimisation measures are measures that are applicable to all products; these measures include the product information, prescription status and pack design. To ensure a positive benefit-risk balance for products associated with serious risks that may not be sufficiently minimised through routine measures, aRMMs may be needed. These aRMMs can range from communication regarding risks supplementary to the product information, to restriction of drug prescribing or dispensing. Previous studies have shown an increase in the number of medicines approved with aRMMs since 2005.
In this PhD project, we have investigated whether the increasing trend in number and proportion of medicines approved with aRMMs continued and whether the new legislation that came into force in 2012 had any impact on the increase. We are also investigating potential predictors of aRMMs, best methods for evaluating the effectiveness of aRMMs, regulatory outcomes of effectiveness evaluations of aRMMs. With our studies, we aim to analyse regulatory decision-making regarding aRMMs and where possible to provide points for attention.
PhD-student: Christel Hoeve (MEB / Erasmus Medical Centre Rotterdam)
Promotor: Prof. Miriam Sturkenboom (Utrecht University)
Co-promotor: Dr. Sabine Straus (MEB / Erasmus Medical Centre Rotterdam)
The focus of this PhD project is on medication errors. Medication errors are defined as an unintended failure in the drug treatment process that leads to, or has the potential to lead to, harm to the patient. The different stakeholders involved in the treatment of patients can all take preventive measures to reduce the risk of medication errors. For regulators some of the potential measures are monitoring of medication error cases in adverse event reports, clear package instructions and labels, or additional risk minimisation measures in the context of the Risk Management Plan (RMP). Within this project we are performing studies on medication errors (in particular vaccination errors) reported to EudraVigilance, the adverse event reporting system of the EMA. In addition, studies on the risk management of medication errors are ongoing. Besides investigating the regulatory part of medication errors, we have also focussed on the adherence of prescribers to recommendations to reduce medication associated hospitalisations. These studies give insight in the adherence of health care professionals to recommendations regarding treatment processes. This may be valuable for regulators when developing new recommendations or risk communications.
PhD-student: Yvette Weesie (Nivel / University of Groningen, faculty of Science and Engineering)
Promotor: Prof. Liset van Dijk (Nivel/University of Groningen , faculty of Science and Engineering) and Prof. François Schellevis (Nivel/ Amsterdam University Medical Center location VUmc)
Co-promotors: Dr. Karin Hek (Nivel) and Dr. Tjard Schermer (Nivel)
Collaborators: Dr. Liesbeth Rook (MEB)
In the past 25 years there has been a 10 to 14 fold increase reported in opioid use in the US and Canada, which can only remotely be explained by an overall increase in the population. These signs are also visible in the Netherlands, where an increase is found in opioid prescriptions like morphine, oxycodone, fentanyl and buprenorphine, although on a smaller scale than in the US. Opioids are prescribed for the treatment of severe pain caused by for example cancer, surgery or an accident. But opioids are increasingly being prescribed for chronic non-cancer pain. There is an ongoing discussion in the literature whether or not the benefits of prescribing (strong-acting) opioids in the long term outweigh the risks in patients with (chronic) non-cancer pain.
In this PhD-study we will try to answer questions like: who initiates the opioid prescription, the GP or the specialist? What is the variation in opioid prescription between GP-practices? What do GPs think about the current developments around opioids? And how do GPs prescribe opioids to patients who are more at risk for adverse effects or fall-related injuries, like the elderly? Can we constantly monitor prescribing of opioids in primary care? The focus of the PhD will be on opioid prescribing in primary care, because general practitioners prescribe a large portion of the opioids and usually manage patients with chronic pain conditions.