We are delighted to announce that Carla Jonker (in February 2022) and Lourens Bloem (in November 2021) have successfully defended their PhD thesis. We congratulate both of them! For this Regulatory Science Magazine we asked them both several questions about the research they conducted.
Carla Jonker obtained her Master’s degree in Biomedical Sciences at the University of Leiden. Following graduation she had different positions at various pharmaceutical companies.
In 2004 she started as Regulatory Project Leader at the Dutch Medicines Evaluation Board. From 2013 she combined this position with a PhD project at the Julius Center for Health Sciences and Primary Care at the University Medical Center Utrecht, University of Utrecht. During her PhD she was an actor in the European Medicines Agency’s Cross Committee Task Force on Registries.
Currently Carla combines her position as Senior Regulatory Project Leader at the Medicines Evaluation Board with her work as National Expert in the Data Analytics and Methodology Task Force at the European Medicines Agency.
Thesis highlight: Carla Jonker
Carla Jonker has successfully defended her PhD thesis at Utrecht University titled: “Rare disease registries: A must for regulatory decision making."
Her supervisors were Prof. Arno Hoes (University Medical Center Utrecht (UMCU)), Prof. Peter Mol (MEB/University Medical Center Groningen (UMCG)) and dr. Marijke van den Berg (PedNet Haemophilia Research Foundation). You can find her thesis here.
What is the most important message from your research?
The aim of this thesis was to investigate the value of registries in the regulatory decision making process for therapies in rare diseases. In general, registries are an important source for information on diseases, including standards of care, safety and effectiveness of treatments and long-term prognosis. The main findings of the studies included in this thesis are:
- In one-third of the newly approved drugs, a registry was required to provide additional data, mostly because of safety concerns. Most of the drugs involved were innovative and orphan drugs.
- In previously untreated patients with severe haemophilia A, using the same recombinant FVIII product, inhibitor development (this is the main adverse effect of factor VIII treatment) proved comparable in a single-arm clinical study and a registry-based study population. Moreover, in the registry-based study, patient numbers and completeness of follow-up were higher.
Rare diseases registries will have an increasingly important role in the regulatory decision process. With the development of targeted therapies, data will be collected in, often small, and single-arm, clinical studies. Registries can serve as a better alternative or as additional tool to provide data on effectiveness and safety.
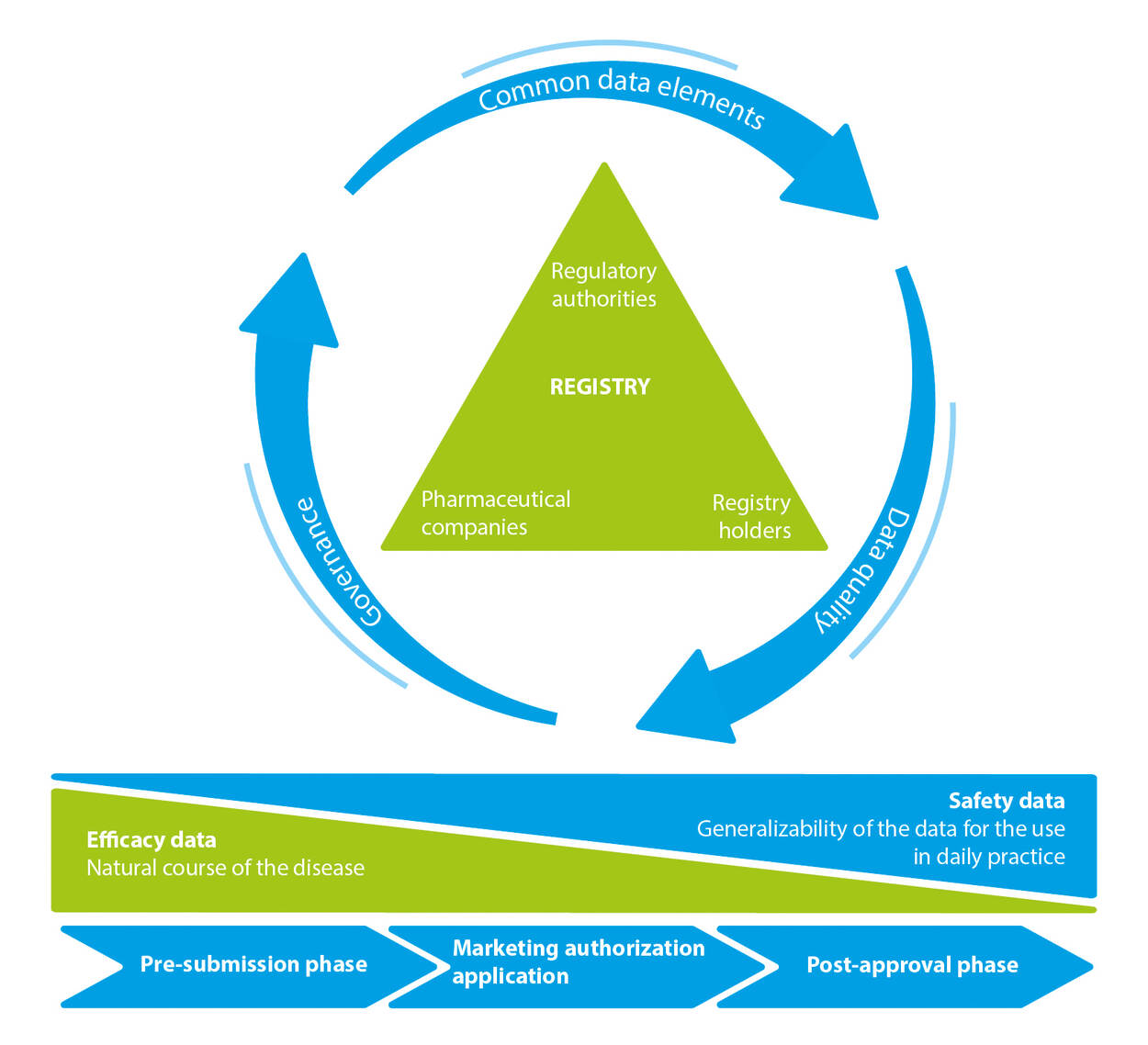
Common data elements > Data quality > Governance
Registry
Regulatory authorities > registry holders > Pharmaceutical companies
Efficacy data Safety data
Natural cause of the disease Generalizability of the data for the use in daily practice
Pre-submission phase > Marketing authorization application > Post-approval phase
What is the impact of your research for the MEB?
In my opinion disease registries should be utilised more often to support regulatory decisions. An ideal framework for use of registry data starts with the regulatory question that defines the data requirements (see Figure 1). Throughout the life cycle of a medicinal product, from the pre-submission to the post-approval phase, data are needed to guide regulators, industry and registry holders (investigators) on the natural course of the disease.
During the drug’s life cycle, other questions become relevant, such as whether the effects observed in a trial population also apply to a “typical” or “average” (real world) patient captured in a disease registry. In addition, questions on long-term safety may only be answered when patients can be followed for many years, not just months. This means that data collection within registries is not static and may require different definitions depending on the regulatory question to be answered. Therefore, regulators, industry and registry holders need to interact early and align expectations, and come to a common understanding of what important questions can be answered with registry-based studies to develop new medicinal products for patients.


Lourens Bloem started his career as a pharmacovigilance assessor at the Medicines Evaluation Board (MEB), after successfully obtaining a Master’s degree in Pharmacy at Utrecht University in 2016. He combined working for the MEB with performing his PhD studies at the Division of Pharmacoepidemiology and Clinical Pharmacology of the Utrecht Institute for Pharmaceutical Sciences at Utrecht University. In 2020, after leaving the MEB he became programme manager Drug Regulatory Science at the Utrecht Science Park.
During and after the finalization of this PhD thesis, Lourens contributed to several research projects of the division for DG SANTE of the European Commission and the National Healthcare Institute (Zorginstituut Nederland), among others. In January 2022, he was appointed assistant professor Clinical Therapeutics at the division. In that capacity, he looks forward to continue performing research in collaboration with the MEB.
Thesis highlight: Lourens Bloem
Lourens Bloem has successfully defended his PhD thesis at Utrecht University titled: “Evidence generation on benefits and risks of medicines and its impact on regulatory and downstream decision-making.”
His supervisors were Prof. Aukje Mantel – Teeuwisse (Utrecht University (UU)) , Prof. Olaf Klungel (UU), dr. Jarno Hoekman (UU) and dr. Menno van der Elst (MEB). His thesis will be publicly available at the end of May. On PubMed, you can find Lourens' publications.
What is the most important message from your research?
For my thesis, we performed studies solely focusing on the regulatory space, as well as studies on the interfaces regulation-health technology assessment (HTA) and regulation-clinical practice. For example, in all three focus areas/interfaces, we performed a study that investigated evidence generation for conditionally authorised medicines. We stress that it is important to have early discussions about and prospectively plan specific obligations for post-authorisation evidence generation. Such discussions should not only be held between applicants and regulatory authorities, but should also be joined by HTA agencies, who may require specific evidence to inform (re-evaluations of) reimbursement decisions. Similarly, the involvement of healthcare professionals in regulatory processes should be increased to ensure understanding of this process of iterative evidence generation and its impact on demonstrable clinical benefit.
Overall, we suggest that the interaction between decision-makers in the medicine lifecycle should be increased. A tool to facilitate this could be a joint medicine-specific evidence generation plan that could guide discussions about unmet medical need, key uncertainties and subsequent requirements for and timing of evidence generation.
What is the impact of your research for the MEB?
We provided various recommendations for regulatory authorities, including the MEB. Apart from the above recommendation for a multi-decision-maker evidence generation plan, we propose among others that the process of post-authorisation evidence generation can be facilitated by ensuring early dialogues about and prospective planning of conditional marketing authorisation (CMA) and specific obligations; by ensuring, where possible, that post-authorisation studies have already started (enrolment) at time of authorisation; and by paying specific attention to formulation and follow-up of these specific obligations. Furthermore, to enhance downstream decision-making processes such as HTA, transparency of the regulatory post-authorisation evidence generation process may be increased through reporting changes in requirements for post-authorisation evidence generation; CMA annual renewal and conversion assessment reports; and increased detail of evidence that led to post-authorisation regulatory actions, including where applicable data source and study design. Lastly, understanding of regulatory decision-making by other decision-makers may be enhanced by better communicating regulatory evidence standards and assessment methods and explaining potential discrepancies with their expectations of evidence standards, such as those underlying clinical benefit scores.
In general, the studies performed for my thesis contributed to the drug regulatory science field that evaluates the functioning of the regulatory system. Moreover, they contributed to my own understanding of the regulatory system and desire to perform further research that hopefully contributes to regulatory and clinical decision-making.